
Our team uses ab initio calculations to explore the mechanisms behind unexplained experimental phenomena and to accelerate the discovery and improvement of materials systems.
ab initio Algorithms Development
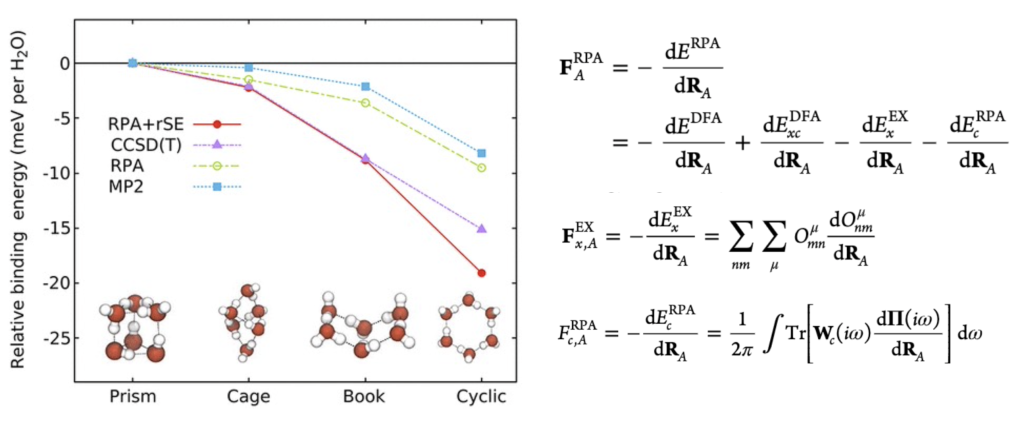
Accuracy is the key for efficient inverse design based on first-principles calculations. As the developers of FHI-aims—one of the most accurate first-principles software packages—our team focuses on developing and implementing the most precise ab initio algorithms within it. [The figure on the left illustrates our implementation of force calculations at the Random Phase Approximation (RPA) level in FHI-aims.]

Machine Learning aids First Principle Software Development
Computational efficiency and scalability are also critical for efficient inverse design based on first-principles calculations. To enable simulations of large material systems (thousands of atoms or more) within seconds, our team focuses on integrating machine learning techniques into modern first-principles software (e.g. FHI-aims, Abinit, QE, VASP, etc.). This approach aims to achieve DFT-level accuracy at significantly reduced computational cost.
Model Development for New Phenomena
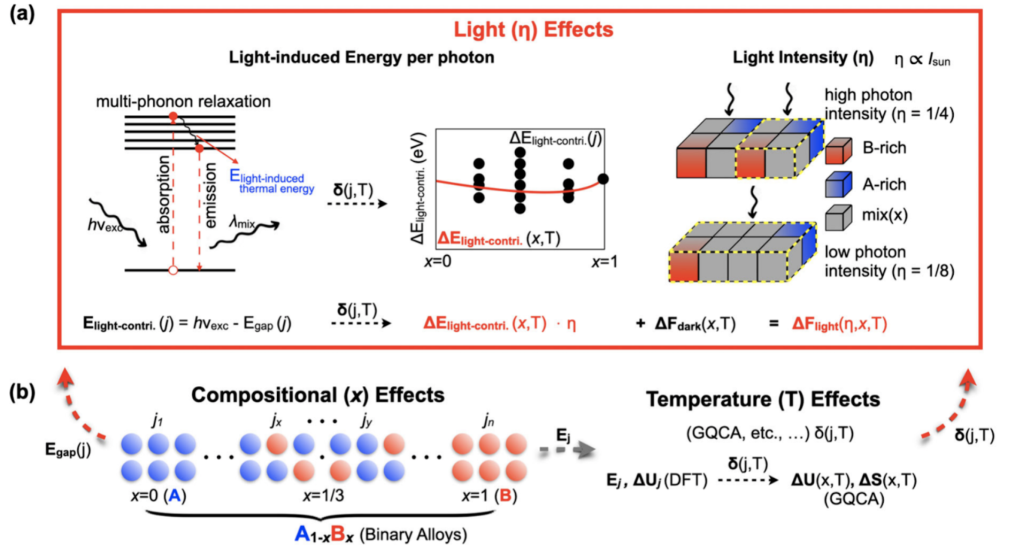
The emergence of numerous new materials systems has revealed many interesting application possibilities based on novel experimental phenomena. However, the underlying mechanisms driving these phenomena often remain unclear to the science community. Our group is dedicated to discovering new physical models to explain and unravel these mechanisms. By further developing and applying such models, we aim to accelerate the discovery of new materials systems through accurate first-principles calculations. [The figure on the left illustrates our new model for simulating light-induced phase segregation in perovskite solar cells. ]
Inverse Design on Various Materials Systems
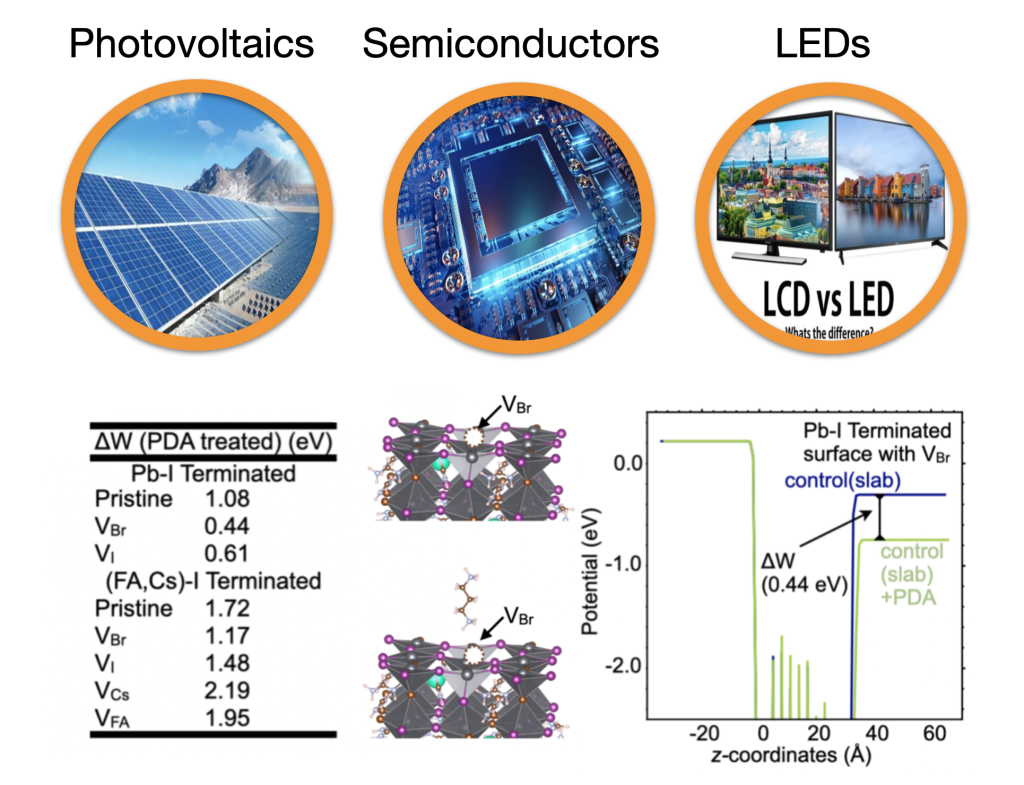
Ultimately, theoretical work must translate into practical applications. Rational materials design benefits significantly from computationally screening large candidate libraries. In our group, we employ highly accurate first-principles calculations to perform inverse design across diverse materials systems – including photovoltaics, semiconductors, LEDs, etc. Leveraging efficient first-principles software and machine learning techniques, we aim to build a comprehensive materials genome database, which will incorporate diverse DFT-calculated properties (structural, electronic, optical, defect-related, transport, etc.). Furthermore, we like to collaborate with experimentalists to explore how machine learning models, trained on this database, can accelerate the discovery of novel materials systems. [The bottom part of the left figure presents our recent Nature-published work utilizing first-principles calculations for inverse design of ligand passivation]